FDA Approval of the Delandistrogene Moxeparvovec-rokl (ELEVIDYS) Gene Therapy for Duchenne’s Muscular Dystrophy Calls into Question the Agency’s Role as a Careful Gatekeeper
Health Letter, November 2023
By Michael T. Abrams, M.P.H., Ph.D.
Senior Health Researcher, Public Citizen’s Health Research Group
Duchenne’s Muscular Dystrophy (DMD) is a catastrophic condition with an aggressive course that leads to skeletal and cardiac muscle degradation in childhood, and to death early in adulthood. In June 2023, the U.S. Food and Drug Administration (FDA) fast-tracked (via accelerated approval) a novel gene therapy for DMD. In testimony before the Cellular, Tissue and Gene Therapies Advisory Committee, and in a follow-up letter to the FDA, Public Citizen’s Health Research Group urged the agency not to approve the gene therapy because evidence for its safety and effectiveness was lacking. After describing the disease and the therapy, this article examines the controversial thinking that led to the accelerated approval decision.
Background on Duchene’s Muscular Dystrophy
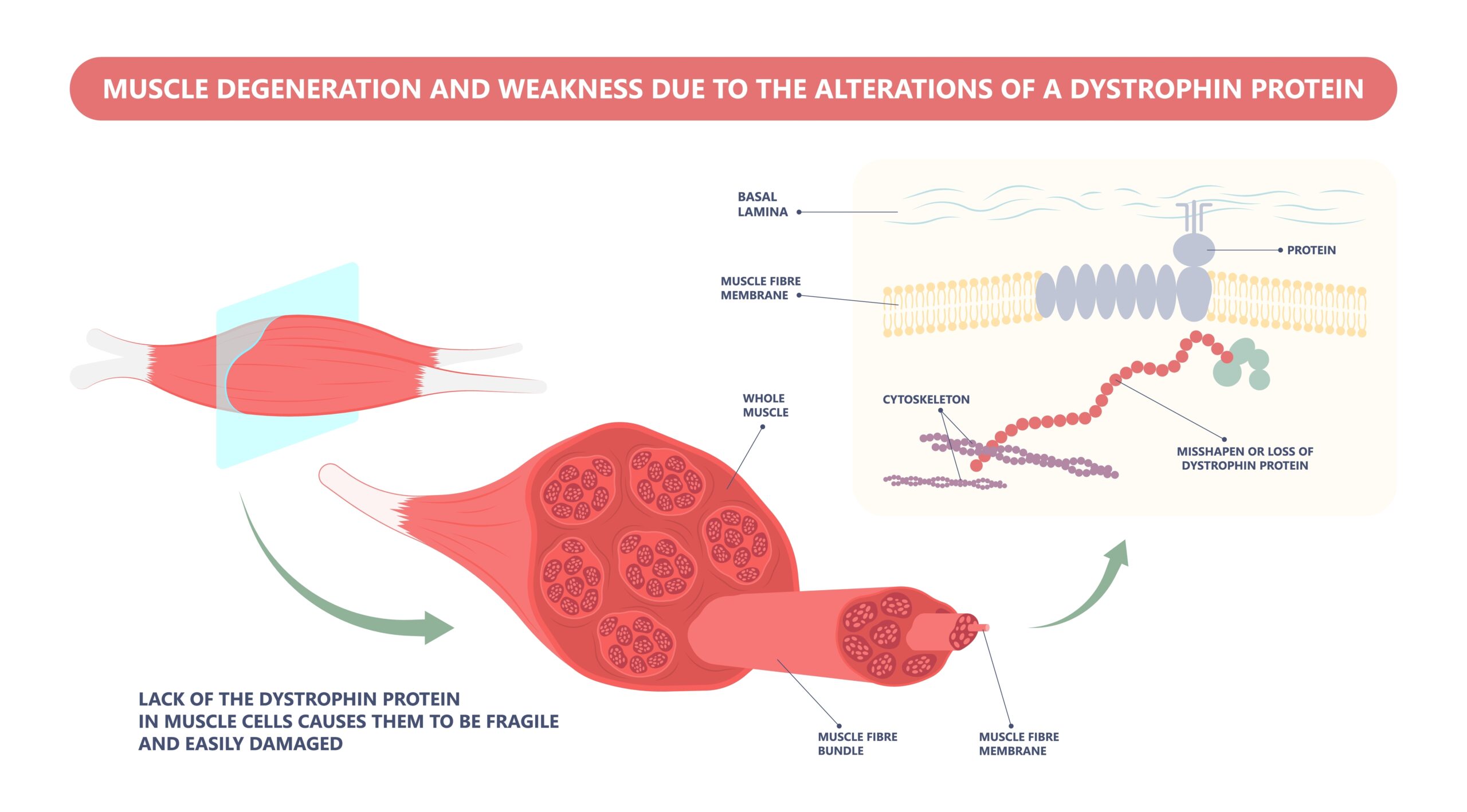
Dystrophin is a complex protein that is believed to act, in part, as a shock absorber, protecting muscle tissue against damage from repeated contraction and extension events. DMD is caused by mutations that silence the dystrophin gene.
Because the dystrophin gene resides on the X chromosome and the disease trait is recessive, males are at high risk for full expression of DMD. The incidence (new cases) of DMD is approximately 1 in 3,600 live male births. Boys with DMD typically develop normally until about 3 years of age, after which they begin to decline. By ages 10-12 years, most require a wheelchair, followed by assisted ventilation at ages 15-20 years. By their late 20s, many patients with DMD die from heart failure.
Becker Muscular Dystrophy (BMD) is a mild form of muscular dystrophy that has inspired the development of a novel gene therapy for DMD. BMD results from mutations that express a very truncated form of the natural dystrophin protein. As compared to patients with DMD, patients with BMD experience milder and delayed loss of muscle function.[1] The BMD-dystrophin might help preserve muscle function in patients with DMD. Additionally, this shortened dystrophin is easier to deliver to patients using existing gene therapy technologies.
Limits of gene therapy for Duchenne’s Muscular Dystrophy
Ideally, a gene therapy for DMD would completely replace the aberrant dystrophin gene with a gene that expresses fully functional dystrophin. The fully functional gene, however, is the largest single gene in the human body, which makes it difficult to deliver into human muscle cells. Because viruses can incorporate bits of genetic code into a patient’s cells, re-engineered viral particles are used to deliver gene therapies for neuromuscular diseases. The adeno-associated virus is a commonly used capsule for carrying genes, but with a capacity well below the size of the full dystrophin gene. Thus, with current viral-delivery technology, gene therapy for DMD is limited to truncated versions of dystrophin, such as the BMD form.
At present, all the FDA-approved, gene-based therapies for DMD have relied on facilitated expression of a truncated dystrophin protein.[2] Since 2016, there have been five therapies, each of which have entered the United States market via the FDA’s “accelerated approval” pathway. The effectiveness of all these treatments is highly questionable.
The latest FDA-approved Duchenne’s muscular dystrophy gene therapy
In 2023, the FDA granted accelerated approval to delandistrogene moxeparvovec-rokl (ELEVIDYS). In contrast to the prior four therapies that worked by “skipping” over bad sections of the patient’s existing gene, the new gene therapy, also known as SRP-9001, replaces a patient’s aberrant dystrophin gene with an externally created dystrophin gene. The FDA approved SRP-9001 to treat DMD in children ages 4-5 years who have confirmed mutations in certain sections of their dystrophin gene.
The gene therapy is administered to patients via an intravenous infusion that lasts 1-2 hours. Subsequently, patients often receive corticosteroids for 60 days. This one-time treatment has been priced by the manufacturer at about $3.2 million. Once administered, a patient can no longer receive a gene therapy administered with an adeno-associated virus, as this could trigger a serious allergic reaction that may lead to death.
FDA’s justification for accelerated approval
A decision memo from Peter Marks, the Director of the Center for Biologics Evaluation and Research at the FDA, explains why the agency approved SRP-9001 over the objections of the agency’s scientists and nearly half of the voting members of the external advisory committee, which voted 8-6 for accelerated approval. Even advisory committee members who supported approval were skeptical. As Marks wrote, “some of the advisors who voted in favor of approval, noting compelling patient testimony and video evidence shown, expressed concern about the strength of the clinical evidence.”
Notably, Marks agreed with the FDA reviewers that the pivotal randomized trial conducted by the company applying for approval failed to show that the gene therapy was effective at slowing disease progression in 20 patients who received the gene therapy, as compared to 21 patients who received a placebo therapy. The trial, in boys ages 4-7 years, followed patients for 48 weeks after treatment. There was no significant motor function change between groups, as measured by a standardized assessment score that ranges from 0 (unable to perform any activities) to 34 (all activities performed normally). Follow-up analysis by the company, however, showed that when the sample was limited to 4-5 year olds (eight in the treatment group, eight in the placebo group), there was a difference of 2.4 points in favor of those receiving the gene therapy. It is well-known by experts in clinical trial methodology that such post-hoc, or “after-the-fact,” analyses are biased toward results that support a hoped-for finding. Unless confirmed in additional studies, post-hoc findings are best disregarded.
In the decision memo, Marks highlighted additional uncertain results for laboratory outcomes pertaining to the 4-5 year old subgroup of patients. The laboratory outcome was the amount of truncated dystrophin protein expressed in the patients receiving the gene therapy. In clinical trials, laboratory outcomes are often less relevant as indicators of treatment effects than clinical outcomes, such as assessments of muscle function in patients with DMD.
As compared to negligible increases in the placebo group, patients receiving the gene therapy averaged 40% increases in the amounts of truncated dystrophin after 12 weeks; however, only three patients showed increases in the amounts of dystrophin of 25% or more. Moreover, other analyses failed to show that the increased dystrophin levels predicted the preservation of motor function one year after the treatment was administered. Thus, there was scant dose-response evidence of the gene therapy’s effectiveness.
In addition to the fact that an adeno-associated virus treatment can be given only once, the gene therapy has risks, including rare but serious liver, heart and skeletal muscle toxicity, and common (≥20%) bouts with nausea, vomiting, pyrexia (fever) and thrombocytopenia (low platelet counts).
Concerns about the FDA’s fast-track approvals of Duchenne’s muscular dystrophy gene therapies
The FDA’s fast-track (accelerated) approval of SRP-9001 and other gene therapies for muscular dystrophy are concerning. Although such approvals may be welcomed by patients, families and the drug companies that make the treatments, they are not decisions based on strong evidence that a gene therapy is safe and effective. In 2016, the first dystrophin-based treatment for DMD, eteplirsen (EXONDYS 51), received accelerated approval based on weak evidence from just 12 patients.[3] Janet Woodcock, then Director of the FDA’s Center for Drug Evaluation and Research, said that the approval of eteplirsen was partly justified because the sponsoring firm “needed to be capitalized” or it would be unable to develop the drug or other treatments for DMD. The sponsoring firm was Sarepta Therapeutics, the same company that manufactures, and now markets, SRP-9001. An August 2023 analysis in Health Affairs discussed the SRP-9001 approval as a prime example of patient advocates (often with industry support) lobbying the agency to approve treatments of dubious to no value; if the FDA does not approve the treatment, it might be seen as impeding innovation.
Conclusions
The accelerated approval of the SRP-9001 gene therapy for early DMD is a troubling example of the FDA granting accelerated approval for a drug with negligible evidence of effectiveness, and serious concerns about adverse effects. There is a critical need for better treatments for DMD. Truncated dystrophins have scientific merit as a potential, albeit partial, treatment. However, the evidence to date—regarding motor function and protein expression—fails to demonstrate that SRP-9001 performs better than a placebo, even in the 4-5 year age group, for which the gene therapy was approved. SRP-9001 should not be used to treat DMD outside of additional and well-designed randomized controlled studies. Moreover, unless additional studies provide compelling clinical evidence of safety and effectiveness, the FDA should rescind the accelerated approval.
References
[1] Darras BT. Duchenne and Becker muscular dystrophy: clinical features and diagnosis. UpToDate. June 22, 2022.
[2] Bendicksen L, Zuckerman DM, Avorn J, et al. The regulatory repercussions of approving muscular dystrophy medications on the basis of limited evidence. Ann Intern Med. 2023;176(9):1251-1256.