Report: Failed Trials, Yet Full FDA Approval of a Duchenne Muscular Dystrophy Gene Therapy
By By Michael T. Abrams, M.P.H., Ph.D.*; Reshma Ramachandran, M.D., M.P.P., M.H.S;** Robert Steinbrook, M.D.*
*Public Citizen’s Health Research Group
** General Internal Medicine, Department of Internal Medicine, and Collaboration for Regulatory Rigor, Integrity, and Transparency. Yale School of Medicine.
Summary
In separate regulatory decisions in 2023 and 2024, the Food and Drug Administration (FDA) approved a novel gene therapy for the rare but deadly childhood-onset disease Duchenne muscular dystrophy. The 2023 approval was limited to children who are 4-5 years of age and able to walk (ambulatory). The 2024 decision expanded that approval to include traditional approval in ambulatory individuals 4 years of age and older with the disease and a confirmed mutation in the DMD gene. The agency also added accelerated approval for wheelchair-dependent individuals 4 years of age and older. Neither approval was supported by favorable FDA scientific reviews or appropriate clinical trial results demonstrating that children with DMD maintained or regained gross motor abilities. Both approvals occurred because an FDA director overruled the agency’s scientific staff. This report describes the two ill-advised approvals, explains why they are examples of actions FDA leaders should avoid in the future, and makes recommendations for how the FDA should change the way it resolves controversial regulatory decisions.
Introduction
On October 29, 2024, Nature Medicine published a report about the two Phase 3 randomized clinical trials of delandistrogene moxeparvovec-rokl (Elevidys) as a gene replacement therapy for Duchenne muscular dystrophy (DMD).[1] That report noted two reasons why the clinical effectiveness of this drug had not been clearly demonstrated. First and foremost, both trials failed to meet their primary endpoints for clinical benefit. Clinical benefit was assessed with a standardized scale measuring gross motor performance (e.g., standing, walking, jumping, and head lifts) trajectory. Second, slightly favorable alternative endpoints — ultimately used by the Food and Drug Administration (FDA) to justify approval — were unverifiable with statistical testing. Moreover, the most important of the alternative outcomes was micro-dystrophin protein levels, a biomarker that recently failed to demonstrate clinical relevance in another DMD gene therapy trial.[2]
The weak clinical evidence notwithstanding, on June 22, 2023, delandistrogene moxeparvovec-rokl became the first FDA-approved gene replacement therapy for DMD, then only for children 4-5 years old who are able to walk (ambulatory).[3] That approval was granted via the FDA’s accelerated approval program.[4] On June 20, 2024, the FDA expanded the approval to include traditional approval in ambulatory individuals 4 years of age and older with the disease and a confirmed mutation in the DMD gene. The agency also added accelerated approval for wheelchair-dependent individuals 4 years of age and older. [5]
The following disturbing statement appeared in the FDA’s clinical review document regarding the expanded approval action:[6]
The CBER [Center for Biologics Evaluation and Research] Director, Dr. Peter Marks, is approving the [application] by overriding the review team’s recommendation…
Accordingly, delandistrogene moxeparvovec-rokl can now be marketed in the United States. The therapy was recently priced at over $3 million per treatment.[7]
DMD is a rare, progressive, and deadly disease with limited treatment options
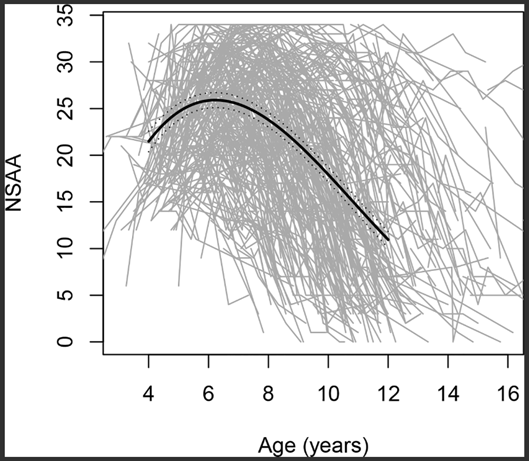
Because they are recessive and X-linked, DMD mutations mostly affect males.[8] The birth incidence in males is approximately 1 in 3,600.[9] Such mutations lead to skeletal and heart-muscle degradation that emerges in early childhood despite other normal muscular-skeletal development. Most individuals born with a DMD genotype lose the ability to walk by early adolescence and die in their thirties.[10] The disease course, however, is heterogeneous, as demonstrated by FDA data showing that the age trends in motor function are quite variable in boys with DMD aged 4-16 years (see figure).[11]
Figure. North Star Ambulatory Assessment (NSAA, gross motor performance scale) score changes with age; 395 individual trajectories included, each represented by a grey line.
Although delandistrogene moxeparvovec-rokl is the first disease-modifying therapy for most DMD cases, the FDA had previously approved four drugs for rarer forms of DMD. The first was the still-questionable drug eteplirsen[12] (Exondys 51), which was developed to treat narrower DMD mutations using antisense oligonucleotides that facilitate targeted “exon skipping” (gene-to-protein expression that avoids mutated regions).[13] Including delandistrogene moxeparvovec-rokl, there are five FDA-approved treatments for DMD, all relying on truncated forms of the muscle-critical protein dystrophin (detailed below). All of these treatments were approved through the FDA’s accelerated approval program. Importantly, although most of these treatments have been used in patients for several years, none of them have demonstrated convincing clinical benefits as compared to treatment with placebo.[14] Moreover, four of the treatments, including delandistrogene moxeparvovec-rokl, are manufactured by one company: Sarepta Therapeutics. Ongoing concerns about Sarepta’s DMD products prompted Dr. Robert Califf, the FDA commissioner when both eteplirsen and delandistrogene moxeparvovec-rokl were approved, to state in 2024, “Sarepta’s like a curse to me.”[15]
Delandistrogene moxeparvovec-rokl’s mechanism of action
Mutations that lead to DMD greatly diminish or silence the expression of a gene that encodes for the protein dystrophin.[16] The protein contains numerous biochemical components that span muscle cell membranes. It also functions as a shock absorber that helps muscle stay resilient despite degradation that may result from repeated contraction-expansion events.
In theory, promoting dystrophin expression in the muscle cells of patients with DMD can mitigate or reverse the disease. This theory inspired the development of delandistrogene moxeparvovec-rokl, whose active ingredient is an engineered RNA sequence that encodes for a truncated form of dystrophin, referred to as “micro-dystrophin.” Micro-dystrophin is about 30% of the size of unmutated dystrophin. The micro-dystrophin coded by delandistrogene moxeparvovec-rokl was designed based on the truncated protein found in patients with a milder form of muscular dystrophy referred to as the Becker type. The truncation of the dystrophin gene was required because the RNA sequence must be packaged into viral capsids for delivery into the body. Such capsids have a capacity limit that is exceeded by the full dystrophin RNA.
Although sophisticated, the bioengineering strategy used to create delandistrogene moxeparvovec-rokl produces a protein that would at best only partially restore dystrophin function throughout the body. Clinical trials have yet to demonstrate that this complex therapy works.
Accelerated approval of 2023
The pivotal trial for the first approval of delandistrogene moxeparvovec-rokl involved 41 ambulatory children with DMD, who ranged in age from 4-7 years, randomized 1:1 to receive the drug or placebo.[17] The primary prespecified clinical outcome was change over 48 months observed on a 17-item standardized scale assessing gross motor performance; there were no significant improvements in performance among the subjects treated with delandistrogene moxeparvovec-rokl as compared with the control group. Because of that negative clinical trial result, the sponsor (Sarepta) sought accelerated approval based on the surrogate endpoint of micro-dystrophin expression in muscle tissue 12 weeks after the gene therapy treatment.
Although micro-dystrophin expression was observed at 12 weeks, micro-dystrophin levels were not correlated with resulting muscle function. Accordingly, the FDA scientific reviewers concluded that the delandistrogene moxeparvovec-rokl trial failed to demonstrate clinical benefit in the treatment of DMD. The 2023 FDA clinical review said this:[18]
These data therefore are insufficient to support expression of ELEVIDYS (sic) micro-dystrophin as a surrogate endpoint “reasonably likely to predict clinical benefit” for Accelerated Approval of ELEVIDYS for even a limited population, such as ambulatory patients age 4 through 5 years with DMD with a confirmed DMD mutation in the DMD gene.
Despite this strong critique, the director of the FDA’s Center for Biologics Evaluation and Research overruled the agency’s scientific reviewers and made the decision to grant accelerated approval in ambulatory 4- and 5-year-olds with DMD. The decision was based mainly on data from 8 of the 41 subjects.[19] The decision further “required as a condition of accelerated approval” that the sponsor complete a second randomized trial, the results of which were anticipated by fall 2023.
Expanded approval in 2024
In an October 30, 2023, press release, Sarepta reported that the second pivotal randomized trial of delandistrogene moxeparvovec-rokl did not achieve its primary endpoint.[20] That placebo-controlled trial involved 125 patients aged 4-7 years who were randomized 1:1 to receive either delandistrogene moxeparvovec-rokl or placebo. The primary outcome was 52-week change on the same muscle function scale score used in the earlier trial. The results were not significant (p=0.25) as they revealed only a 0.65-point average difference between drug and placebo on the standardized gross motor functional scale (which has a maximal range of 34 points).
Delandistrogene moxeparvovec-rokl’s run as an FDA-approved drug should have ended there. However, the manufacturer instead highlighted secondary outcomes that they claimed yielded “robust, statistically significant results…[that] support an efficacy supplement” to the 2023 approval.
Reviewing this new evidence, the FDA scientific reviewers were pointedly critical. Specifically, two of the key secondary outcomes were time to rise from lying on the floor and the 10-meter walk/run tests. Subjects who received delandistrogene moxeparvovec-rokl performed, on average, about 0.5 seconds better than placebo recipients on these tasks, which at baseline took about 3.5 and 4.9 seconds, respectively. The FDA reviewers said these results, which were neither prespecified nor statistically adjusted for the repeated analyses of the data, “cannot support effectiveness of ELEVIDYS.” Moreover, the FDA reviewers said the results were “misleading and cannot guide any stakeholder — including patients, family members and caregivers, and prescribers — in making informed decisions about the potential benefits of treatment with ELEVIDYS.”[21]
Nonetheless, for a second time and despite the negative reviews from FDA scientists,[22] the CBER Director in June 2024 overruled his staff and green-lighted delandistrogene moxeparvovec-rokl.[23] The second approval, as discussed above, hardened and broadened the approval to include traditional approval in ambulatory individuals 4 years or age and older with the disease and a confirmed mutation in the DMD gene. The second approval also added accelerated approval for wheelchair-dependent individuals 4 years of age and older.
In his 2024 decision memo the CBER Director based his decision largely on the surrogate indicator of correlation between levels of the micro-dystrophin protein and the time to rise from lying on the floor data. That correlation, however, was neither statistically significant (p=0.1388) nor visually (graphically) persuasive because the slope was shallow (-0.012 seconds for every percent change in micro-dystrophin level in the muscle). Moreover, many of the data points fell far off the derived regression line.[24] In considering the micro-dystrophin dose-response data, FDA scientists had specifically written that the results should be “interpreted with caution” because only 25% of the subjects in the second pivotal trial were included in this analysis. Accordingly, although there is a scientific rationale for micro-dystrophin gene insertion as a treatment for DMD, trials have not demonstrated that the gene therapy leads to any clinical benefit. In fact, a recent Phase 3 trial of a DMD gene therapy developed by Pfizer yielded “significant amounts” of micro-dystrophin expression in the muscles of DMD patients, but no corresponding clinical improvement. Pfizer reportedly has discontinued development of their therapy.[25]
FDA documents show that two other supervisory-level FDA staff, both in CBER’s Office of Clinical Evaluation, disagreed with the decision to approve delandistrogene moxeparvovec-rokl. They jointly wrote that Sarepta’s latest data on delandistrogene moxeparvovec-rokl [26]
- do not verify the clinical benefit of Elevidys in ambulatory boys aged 4-5 years (i.e., the group for which accelerated approval was granted by Center Director override of the review team and senior CBER leadership recommendation),
- do not demonstrate the benefit of Elevidys in ambulatory patients greater than 5 years of age, or
- do not demonstrate the benefit of Elevidys in non-ambulatory patients of any age.
Persons directly affected by DMD have expressed similar concerns. One parent wrote in September 2024 that they decided not to seek delandistrogene moxeparvovec-rokl treatment for their child because of concerns about safety and effectiveness.[27] Another parent, in a video posted on the internet, criticized Sarepta for developing a treatment that was inadequate and likely to soon become obsolete. Sarepta Therapeutics was alleged to have responded to that latter criticism by threatening to withdraw its financial support from a parent support group hosting the video, which led the group to remove the delandistrogene moxeparvovec-rokl video-critique from its website.[28]
Conclusion
The FDA’s approval of delandistrogene moxeparvovec-rokl as a gene therapy for DMD resulted from the decision by a center director to overrule the agency’s scientific staff who were assigned to evaluate the treatment. Neither of the two pivotal trials of delandistrogene moxeparvovec-rokl met their primary clinical endpoints. Secondary endpoints (e.g., 10-meter walk/run tests) and the indirect surrogate indicator of micro-dystrophin levels, indicators used to salvage the delandistrogene moxeparvovec-rokl application, were themselves controversial. Accordingly, we believe the FDA leadership has set a dangerous precedent that fails to protect the public from exposure to a physically and financially costly therapy that is ineffective.
One response to the FDA’s decision is to conclude that lax approval standards are the problem and the remedy should be stronger approval standards and post-marketing requirements.[29] Although we agree that both stronger approval standards and post-marketing requirements are needed, the FDA’s internal procedures should also be revised to make it far more difficult for individual officials in leadership positions to overrule the consensus evaluation of agency scientists assigned to a regulatory decision.
Califf, the FDA Commissioner at the time of the two approval decisions for delandistrogene moxeparvovec-rokl, has said that critics of the decision have a “very simplistic view of clinical evidence” regarding the challenges of finding treatments for rare and serious conditions.[30] We disagree with that assessment. The approval of delandistrogene moxeparvovec-rokl should prompt reforms within the FDA to improve its decision-making process for new drugs, regardless of whether the drugs are for rare or more common conditions.
In an interview before leaving the agency, Califf also addressed the decision by noting that families with such rare and dire diseases were rationally desperate for any hope, and he as a political appointee was also hesitant to overrule the determinations of career FDA staff.[31] These considerations, although important, are insufficient justifications for the FDA’s approval of delandistrogene moxeparvovec-rokl.
[1] Mendell JR, Muntoni F, McDonald CM, et al. AAV gene therapy for Duchenne muscular dystrophy: the EMBARK Phase 3 randomized trial. Nat Med. 2024 Oct 9. doi: 10.1038/s41591-024-03304-z. Epub ahead of print. PMID: 39385046.
[2] Mast J, Feuerstein A. Perplexing results from Duchenne muscular dystrophy trial raise questions about gene therapies. STAT+. October 21, 2024.
[3] U.S. Food and Drug Administration. News release: FDA approves first gene therapy for treatment of certain patients with Duchenne muscular dystrophy. June 22, 2023. https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-treatment-certain-patients-duchenne-muscular-dystrophy. Accessed January 8, 2025.
[4] U.S. Food and Drug Administration. Accelerated approval program. December 24, 2024. https://www.fda.gov/drugs/nda-and-bla-approvals/accelerated-approval-program. Accessed January 8, 2025.
[5] U.S. Food and Drug Administration. News release: FDA expands approval of gene therapy for patients with Duchenne muscular dystrophy. June 20, 2024. https://www.fda.gov/news-events/press-announcements/fda-expands-approval-gene-therapy-patients-duchenne-muscular-dystrophy. Accessed January 8, 2025.
[6] U.S. Food and Drug Administration. BLA integrated clinical and clinical pharmacology review memorandum, efficacy supplement for delandistrogene moxeparvovec-rokl (STN125781/34). June 18, 2024. https://www.fda.gov/media/179486/download?attachment. Accessed January 8, 2025.
[7] Bendicksen L, Kesselheim AS, Rome BN. Spending on targeted therapies for Duchenne muscular dystrophy. JAMA. 2024;331(13):1151–1153.
[8] Darras BT. Duchenne and Becker muscular dystrophy: clinical features and diagnosis. UpToDate. September 30, 2024.
[9] Venugopal V, Pavlakis S. Duchenne Muscular Dystrophy. 2023 Jul 10. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 29493971.
[10] U.S. Food and Drug Administration. Briefing document regarding delandistrogene maxeparvovec BLA#125781/00. Cellular, Tissue and Gene Therapies Advisory Committee Meeting. May 12, 2023. https://www.fda.gov/media/168021/download. Accessed January 8, 2024.
[11] Muntoni F, Domingos J, Manzur AY, et al. Categorizing trajectories and individual item changes of the North Star Ambulatory Assessment in patients with Duchenne muscular dystrophy. PLoS One. 2019;14(9):e0221097.
[12] ClinicalTrials.gov. Study of Eteplirsen in DMD patients (PROMOVI), NCT02255552. Last updated January 21, 2021. https://www.clinicaltrials.gov/study/NCT02255552?tab=results#results-overview. Accessed January 8, 2025.
[13] Quemener AM, Bachelot L, Forestier A, et al. The powerful world of antisense oligonucleotides: from bench to bedside. Wiley Interdiscip Rev RNA. 2020;11(5):e1594.
[14] Bendicksen L, Zuckerman DM, Avorn J, et al. The regulatory repercussions of approving muscular dystrophy medications on the basis of limited evidence. Ann Intern Med. 2023;176(9):1251-1256.
[15] Trang B. ‘Sarepta’s like a curse on me’: FDA commissioner dismisses controversy over Elevidys. STAT+. September 11, 2024.
[16] U.S. Food and Drug Administration. Briefing document regarding delandistrogene maxeparvovec BLA#125781/00. Cellular, Tissue and Gene Therapies Advisory Committee Meeting. May 12, 2023. https://www.fda.gov/media/168021/download. Accessed January 8, 2024.
[17] Ibid.
[18] U.S. Food and Drug Administration. BLA clinical review memo for delandistrogene moxeparvovec-rokl (STN 125781/0). June 22, 2023. https://www.fda.gov/media/170230/download?attachment. Access January 8, 2025.
[19] Marks P. Center director decisional memo, BLA 125781, ELEVIDYS (delandistrogene moxeparvovec-rokl). Undated. https://www.fda.gov/media/169707/download?attachment. Accessed January 8, 2025.
[20] Sarepta Therapeutics announces topline results from EMBARK, a global pivotal study of ELEVIDYS gene therapy for Duchenne muscular dystrophy. October 30, 2023. https://investorrelations.sarepta.com/node/22976/pdf. Accessed January 8, 2025.
[21] U.S. Food and Drug Administration. BLA integrated clinical and clinical pharmacology review memorandum, efficacy supplement for delandistrogene moxeparvovec-rokl (STN125781/34). June 18, 2024. https://www.fda.gov/media/179486/download?attachment. Accessed January 8, 2025.
[22] U.S. Food and Drug Administration. Supplemental approval letter for BLA 125781/34 (delandistrogene moxeparvovec-rokl. June 20, 2020. https://www.fda.gov/media/179484/download?attachment. Accessed January 8, 2025.
[23] Marks P. Center director decisional memo for BLA 125781/Amendment 34, ELEVIDYS (delandistrogene moxeparvovec-rokl). Undated. https://www.fda.gov/media/179485/download?attachment. Accessed January 8, 2025.
[24] U.S. Food and Drug Administration. BLA integrated clinical and clinical pharmacology review memorandum, efficacy supplement for delandistrogene moxeparvovec-rokl (STN125781/34) . June 18, 2024. https://www.fda.gov/media/179486/download?attachment. Accessed January 8, 2025.
[25] Mast J, Feuerstein A. Perplexing results from Duchenne muscular dystrophy trial raise questions about gene therapies. STAT+. October 21, 2024.
[26] Fashoyin-Aje LA, Verdun N. Office Director Memorandum, Office of Clinical Evaluation, Summary of regulatory decision on sBLA 12578/43. Undated. https://www.fda.gov/media/179487/download. Accessed January 8, 2025.
[27] Werner MC. First opinion: Breakthrough therapies have given Duchenne muscular dystrophy families like mine hope — and new fears. STAT+. September 9, 2024.
[28] Feuerstein A. Sarepta demanded Duchenne patient advocacy group censor video critical of the company. STAT+. July 29, 2024.
[29] Bendicksen L, Zuckerman DM, et al. The regulatory repercussions of approving muscular dystrophy medications on the basis of limited evidence. Ann Intern Med. 2023;176(9):1251-1256.
[30] Trang B. ‘Sarepta’s like a curse on me’: FDA commissioner dismisses controversy over Elevidys. STAT+. September 11, 2024.
[31] Herper M. Leaving FDA, Califf is unapologetic—and warns of staff departures. STAT. January 8, 2025.